



2. 广西中医药大学, 广西海洋药物重点实验室, 广西南宁 530200
2. Guangxi Key Laboratory of Marine Drugs, Guangxi University of Chinese Medicine, Nanning, Guangxi, 530200, China
细胞外信号调节激酶2(Extracellular Signal-Regulated Kinase 2,ERK2)位于丝裂原活化蛋白激酶(Mitogen-Activated Protein Kinases,MAPK)信号通路下游,是细胞外信号调节激酶(ERK)家族成员之一[1]。作为MAPK信号通路的核心承接分子,ERK2参与调控细胞黏附、细胞周期进程、细胞迁移、细胞存活及分化、细胞增殖和转录等大量生理活动,与恶性肿瘤的产生和发展有着密不可分的关系[2]。因此,靶向ERK2小分子抑制剂的设计研发或将为恶性肿瘤治疗提供新型有效的方案。
ERK2被磷酸化后将进入细胞核或其他激活位点,能在乳腺癌[3]、肝癌[4, 5]和宫颈癌[6]等多个类型的肿瘤组织中再次被激活或高水平表达。直接靶向抑制ERK2活性不仅有望成为恶性肿瘤治疗的有效手段,而且能够与MAPK上游激酶抑制剂产生协同效应,为克服药物的耐药性提供新的解决方案[7, 8]。例如,在经典的肿瘤信号传导通路RAS-RAF-MEK-ERK[9]中,因上游靶点(RAF、MEK等)突变而导致获得性耐药的情况时有发生[7-9],其耐药机制大都依赖于ERK2信号转导的重新激活[10, 11],因此联合靶向ERK2的药物可缓解甚至解决上游靶点的耐药性问题。尽管ERK2抑制剂显示出良好的治疗前景,但是目前还没有靶向ERK2的药物获批上市,许多化合物均处于临床试验或临床前研究阶段,例如VTX-11e治疗胰腺癌已进入2期临床研究,且已被证实可诱导胰腺癌细胞系中的细胞凋亡[12];SCH772984治疗结直肠癌已进入临床研究[13];BVD-523治疗转移性葡萄膜黑色素瘤、急性骨髓性白血病和骨髓增生异常综合征已进入2期临床研究,其联合Hydroxychloroquine治疗晚期胃肠道恶性肿瘤也已开展临床研究[14-16];此外,MK-8353[14]、GDC-0994[14]、LY3214996[17]、JSI-1187[18]、ASN007[19]和ASTX029[20]等靶向ERK2的化合物均已引起人们的广泛关注。
北部湾由于其特殊的地理位置,具有独特的海洋生态系统,蕴藏着丰富的待开发海洋生物资源。Wang等[21]系统地总结了近十年从北部湾新发现的477种海洋天然产物及其生物活性,表明北部湾药物资源蕴藏丰富,可进一步发掘利用。本实验室前期基于北部湾次级代谢产物及化学合成已建立了小分子化合物库,因此本研究拟通过体外活性试验从中筛选出作用于人源ERK2的小分子抑制剂(苗头小分子化合物),并通过分子对接初步阐明苗头小分子化合物与ERK2间的结合模式,确定配体在受体活性位点上的结合构象,从而揭示关键的分子间相互作用,为靶向ERK2的药物设计提供新思路。
1 材料与方法 1.1 仪器和试剂5920R高速大容量冷冻离心机(德国Eppendorf公司),VICTOR NIVO酶标仪(Perkin Elmer公司),VCX-750超声破碎仪(美国Sonics公司),Bio-rad Mini-Protean垂直电泳仪(美国BIO-RAD公司),VILBE Quantum CX5凝胶成像仪(法国VILBER BIO IMAGING公司),异丙基硫代半乳糖苷(IPTG,北京索莱宝科技有限公司),Ni Sepharose 6 Fast Flow Purification Resin(美国Cytiva生物科技有限公司),BCA蛋白浓度测定试剂盒(北京索莱宝科技有限公司),ADP-GloTM Kinase Assay试剂盒[普洛麦格(北京)生物技术有限公司]。宿主菌大肠杆菌(Escherichia coli)Rosetta2(DE3)购于西格玛奥德里奇(上海)贸易有限公司。
1.2 质粒构建与蛋白表达依据Heightman等[22]的研究方法确定重组质粒(pET30-ERK21-360)的构建方案,在ERK2(Uniprot ID:P28482,共360个氨基酸,分子量为41.4 kD)的C端引入多组氨酸标签(His-tag),并连接在pET30a载体上,委托武汉淼灵生物科技有限公司构建重组质粒(pET30-ERK21-360)。将重组质粒转入DH5α感受态细胞中进行克隆复制,提取质粒测序并验证重组质粒无误后,将其通过热激法转入Rosetta2(DE3)大肠杆菌感受态细胞诱导表达。在不同IPTG浓度、温度及诱导时间条件下收集菌体并置于冰浴中超声破碎,通过SDS-PAGE凝胶电泳判断蛋白的表达量并据此优化表达条件。将上清液通过镍(Ni)螯合柱完成蛋白纯化,用BCA蛋白浓度测定试剂盒测定纯化后的蛋白浓度。
1.3 体外激酶活性测试苗头小分子化合物对ERK2的活性抑制作用通过ADP-GloTM Kinase Assay试剂盒进行评价。以纯化后的蛋白作为待测样品,根据ADP-GloTM Kinase Assay试剂盒操作手册进行活性测定。首先,将2 μL纯化后的蛋白和1 μL通过梯度稀释(待测化合物初始浓度为5 mmol/L,使用倍比稀释法稀释11个浓度梯度)后的候选化合物依次添加至384孔板中,室温静置2 min后再添加2 μL浓度为100 μmol/L的ATP到384孔板中,室温下静置20 min;然后加入5 μL ADP-Glo试剂,室温下孵育40 min;最后加入10 μL Detection试剂,避光孵育30 min。使用酶标仪读取光信号,并通过GraphPad Prism软件拟合得到化合物浓度与吸光度的响应曲线及半抑制浓度(IC50)值。
1.4 分子对接计算流程 1.4.1 蛋白的预处理ERK2三维晶体结构(PDB ID:4QTE)来自RCSB蛋白质数据库(http://www.rcsb.org)。通过Schrödinger Maestro 11.9软件的Protein Preparation模块对受体蛋白结构进行预处理,设小分子化合物几何坐标的质心为活性口袋中心,将其周围20 Å的立方体设为活性对接口袋。
1.4.2 小分子化合物库的准备小分子化合物库主要来自本课题组构建的北部湾次级代谢产物及化学合成化合物库。利用Schrödinger Maestro 11.9软件的Ligand Preparation模块建立化合物三维结构数据库,模拟pH值为5.0-9.0时小分子化合物可能存在的质子化状态。
1.4.3 分子对接流程及打分采用Schrödinger Maestro 11.9软件的Glide模块进行分子对接,对接精度设置为标准模式(Standard Precision, SP),化合物的分子对接打分采用Glide gscore(Gscore)。考虑溶剂化对结合能力的影响,采用基于对接构型的分子力学-广义玻恩表面积(MM-GBSA)模块计算对接构象的结合自由能预测值。
2 结果与分析 2.1 蛋白的表达与优化根据菌液的电泳结果逐步优化诱导条件[图 1(a)],结果确定蛋白成功表达,且使用20 mL培养基小量表达时,蛋白表达量随温度和时间的增加而上升。然而,若使用IPTG终浓度为0.5 mmol/L、25℃诱导12 h作为诱导条件,在使用200 mL培养基放大条件表达时将出现超声破碎后无法获得澄清液体的情况,判断是由于蛋白表达速度过快而产生了包涵体。为了确保可溶性目的蛋白的高表达,综合比较后确定诱导条件为IPTG终浓度0.1 mmol/L,20 ℃低温诱导9 h。最后,采用200 mL培养基放大条件完成目的蛋白的诱导表达,经过超声破碎及Ni层析柱纯化后获得的蛋白浓度为(1.30±0.41) mg/mL[图 1(b)]。

|
| Lane 1-3, induced at 15, 20, 25 ℃ for 9 h with 0.5 mmol/L IPTG; Lane 4, benchmark pre-stained protein ladder (kD); Lane 5, control of un-induced supernatant (Control check, CK); Lane 6-9, induced time of 3, 6, 9 and 12 h at 20 ℃ with 0.5 mmol/L IPTG; Lane ①, benchmark pre-stained protein ladder (kD); Lane ②, control of un-induced supernatant (Control check, CK); Lane ③, supernatant (SN); Lane ④-⑥, elution with 125, 250 and 500 mmol/L imidazole.The arrows indicate the target protein, ERK2. 图 1 ERK2表达条件优化及纯化的SDS-PAGE鉴定 Fig.1 SDS-PAGE identification of ERK2 expression condition optimization and purification |
2.2 小分子化合物库的分析
本课题组所构建的次级代谢产物库有111个结构明确的天然化合物,包含2, 4-吡咯烷二酮[23]、averantin类似物[24]和carneusins[25]等结构新颖的次级代谢产物;化学合成化合物库目前有吡啶脲类化合物17个[26]。通过对化合物分子量(MW)及脂水分配系数(AlogP)的计算,发现当前化合物库总体成药性较好,大部分化合物均满足成药五规则。其中,化合物平均分子量和AlogP分别为(353.3±100.3) Da和(2.4±1.5)(图 2)。根据化合物的成药五规则,剔除不符合条件的化合物23个,利用分子对接预测了剩余105个化合物与ERK2的结合活性,根据分子对接打分从中选择18个化合物进行生物活性验证。

|
| 图 2 化合物库的性质预测(n=128) Fig.2 Property profiling of compound dataset(n=128) |
2.3 激酶体外抑制活性及其作用模式预测
从2.2节所得的18个化合物中筛选出对ERK2抑制活性较强的部分化合物(记作化合物1-5),其测试结果如图 3所示。化合物1-5作用于ERK2的IC50值分别为(4.17±1.82)、(13.39±0.93)、(27.85±2.55)、(19.14±1.02)和(8.55±3.72) μmol/L,其分子对接打分结果及通过MM/GBSA预测的结合自由能如表 1所示,从中可以看出这5个化合物的分子对接打分绝对值均高于6 kcal/mol,结合自由能预测绝对值均高于38 kcal/mol,说明此时的受体与配体具备较强的结合能力。

|
| 图 3 化合物1-5对ERK2的IC50 Fig.3 IC50 of compound 1-5 on ERK2 |
| 化合物 Compound |
结构 Structure |
半抑制浓度(IC50)/(μmol/L) Half-maximal inhibitory concentration(IC50)/(μmol/L) |
分子对接打分 Gscore/(kcal/mol) |
结合自由能预测值/(kcal/mol) Predicting value of binding free energy/(kcal/mol) |
| 1 | 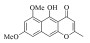
|
4.17±1.82 | -6.628 | -49.96 |
| 2 | 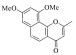
|
13.39±0.93 | -8.626 | -38.88 |
| 3 | 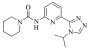
|
27.85±2.55 | -7.067 | -43.03 |
| 4 | 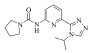
|
19.14±1.02 | -7.019 | -44.35 |
| 5 | 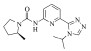
|
8.55±3.72 | -6.164 | -38.35 |
尽管化合物 1与 2是从北部湾真菌代谢产物中分离得到,但二者均为已知化合物。其中,化合物 1为6-甲氧基红镰霉素B,化合物 2为异红镰霉素,它们也是传统中药材决明子中的主要化学成分之一[23, 27]。虽然两者化学结构类似,在特定条件下结构可以互相转化[24],但是不同的作用模式很可能导致两者活性有差异。图 4的分子对接姿势表明化合物 1可以与结合口袋周围的LYS-54、GLN-105和MET-108残基分别形成氢键,而化合物 2则缺失与GLN-105的氢键作用,这也许是化合物 2活性略低于化合物 1的直接原因。

|
| Yellow dash lines represent the H-bond interaction (Å). 图 4 化合物 1、2与ERK2的分子对接姿势 Fig.4 Molecular docking pose of compounds 1, 2 with ERK2 |
化合物 3-5是实验室内部合成的化合物[26],它们母核结构类似,分子对接结果显示该类化合物与ERK2作用模式相同,能够与LYS-54及MET-108形成氢键作用[图 5:(a)-(c)]。该类化合物与已报道的细胞凋亡信号调节激酶1(ASK1)抑制剂结构高度相似[图 5(d)][25]。由于ERK2与ASK1同源,催化域的氨基酸序列比对显示两者序列相似度高达33%(图 6)。ERK2的分子对接结果中复合物结构与ASK1晶体结构(6XIH)高度相似的结合模式也表明当前对接构型的合理。

|
| Yellow dash lines represent the H-bond interaction (Å). 图 5 化合物 3-5与ERK2的分子对接姿势[(a)-(c)]与含有抑制剂的ASK1晶体结构(d)的对比 Fig.5 Comparison between molecular docking pose of compounds 3-5 with ERK2 [(a)-(c)] and crystal structure (d) of ASK1 containing inhibitors |

|
| The sequence of the catalytic domain of ERK2 is the first line (UNIPROT ID P28482) and the sequence of the catalytic domain of ASK1 is the second line (UNIPROT ID Q99683). 图 6 ERK2与ASK1催化域的氨基酸序列比对 Fig.6 Sequences comparison of the catalytic domains of ERK2 and ASK1 |
3 讨论 3.1 化合物1-5与ERK2的作用模式
本研究通过体外激酶活性测试筛选出5个对ERK2具有较高抑制活性的化合物,且具有相同的作用模式,其中最主要的氢键作用氨基酸为赖氨酸(LYS-54)和甲硫氨酸(MET-108)。靶向ERK2的小分子也很有可能会抑制其他激酶,这也是激酶抑制剂普遍面临的选择性难题。值得注意的是,ERK2的门控位点是谷氨酰胺(GLN-105),该氨基酸在激酶家族门控位点中出现的概率不足2%(仅8/516个)[28]。因此,在小分子结构优化时可适当引入与GLN-105作用的片段,从而提高化合物的选择性[29]。
最近,有研究报道ERK2存在变构结合位点[30],考虑到红镰霉素类化合物结构简单,其可能的结合位点还需进一步的确认,可尝试通过氨基酸定点突变或者对小分子进行结构修饰来测试化合物活性的变化。
3.2 化合物的分子对接打分与活性间关系化合物的分子对接打分旨在通过评估化合物与受体蛋白之间的相互作用来预测生物活性。当前5个化合物的分子对接打分与体外活性数据[lg(IC50)]无强相关性,其可能的原因:(1)化合物作用模式的一致性。虽然当前分子对接结果显示5个化合物均与LYS-54、GLN-105和MET-108相互作用,但是实际的作用模式有待进一步验证。(2)分子对接打分函数的合理性。当前的打分函数主要依靠半经验方法,在本研究中的普适性存疑,相比分子对接打分,考虑溶剂化效应的MM-GBSA结合自由能预测表现更优(表 1),但数值上却整体严重高估结合自由能。因此,需要借助基于微扰的分子动力学模拟高精度地计算化合物与ERK2的结合自由能,再对化合物的活性进行验证与评估[31]。需要注意的是,当前活性化合物个数较少,模型打分的准确性还有待进一步验证。
4 结论作为MAPK信号通路的核心承接分子,ERK2与恶性肿瘤的产生和发展有着密不可分的关系。本研究以ERK2为靶标,通过生物活性筛选获得5个具有微摩尔级抑制活性的苗头小分子化合物,并通过分子对接初步探讨了这些化合物与ERK2的可能作用模式,为后续靶向ERK2小分子抑制剂的结构修饰提供了优化思路,也为靶向ERK2抑制剂设计提供了参考。
| [1] |
JR ROSKOSKI R. ERK1/2 MAP kinases: structure, function, and regulation[J]. Pharmacological Research, 2012, 66(2): 105-143. DOI:10.1016/j.phrs.2012.04.005 |
| [2] |
KARNOUB A E, WEINBERG R A. Ras oncogenes: split personalities[J]. Nature Reviews Molecular Cell Biology, 2008, 9(7): 517-531. DOI:10.1038/nrm2438 |
| [3] |
ZHANG D P, LI X W, YAO Z M, et al. GABAergic signaling facilitates breast cancer metastasis by promoting ERK1/2-dependent phosphorylation[J]. Cancer Letters, 2014, 348(1/2): 100-108. |
| [4] |
SHILO A, BEN HUR V, DENICHENKO P, et al. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development[J]. RNA, 2014, 20(4): 505-515. DOI:10.1261/rna.042259.113 |
| [5] |
WANG C, JIN H J, GAO D M, et al. Phospho-ERK is a biomarker of response to a synthetic lethal drug combination of sorafenib and MEK inhibition in liver cancer[J]. Journal of Hepatology, 2018, 69(5): 1057-1065. DOI:10.1016/j.jhep.2018.07.004 |
| [6] |
LI X W, TUERGAN M, ABULIZI G. Expression of MAPK1 in cervical cancer and effect of MAPK1 gene silencing on epithelial-mesenchymal transition, invasion and metastasis[J]. Asian Pacific Journal of Tropical Medicine, 2015, 8(11): 937-943. DOI:10.1016/j.apjtm.2015.10.004 |
| [7] |
VILLANUEVA J, VULTUR A, LEE J T, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K[J]. Cancer Cell, 2010, 18(6): 683-695. DOI:10.1016/j.ccr.2010.11.023 |
| [8] |
CORCORAN R B, EBI H, TURKE A B, et al. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib[J]. Cancer Discovery, 2012, 2(3): 227-235. DOI:10.1158/2159-8290.CD-11-0341 |
| [9] |
CAUNT C J, SALE M J, SMITH P D, et al. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road[J]. Nature Reviews Cancer, 2015, 15(10): 577-592. DOI:10.1038/nrc4000 |
| [10] |
LITTLE A S, SMITH P D, COOK S J. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors[J]. Oncogene, 2013, 32(10): 1207-1215. DOI:10.1038/onc.2012.160 |
| [11] |
LONG G V, FUNG C, MENZIES A M, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma[J]. Nature Communications, 2014, 5: 5694. DOI:10.1038/ncomms6694 |
| [12] |
ARONOV A M, TANG Q, MARTINEZ-BOTELLA G, et al. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control[J]. Journal of Medicinal Chemistry, 2009, 52(20): 6362-6368. DOI:10.1021/jm900630q |
| [13] |
MORRIS E J, JHA S, RESTAINO C R, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors[J]. Cancer Discovery, 2013, 3(7): 742-750. DOI:10.1158/2159-8290.CD-13-0070 |
| [14] |
JR ROSKOSKI R. Targeting ERK1/2 protein-serine/threonine kinases in human cancers[J]. Pharmacological Research, 2019, 142: 151-168. DOI:10.1016/j.phrs.2019.01.039 |
| [15] |
WU J, LIU D, OFFIN M, et al. Characterization and management of ERK inhibitor associated dermatologic adverse events: analysis from a nonrandomized trial of ulixertinib for advanced cancers[J]. Investigational New Drugs, 2021, 39(3): 785-795. DOI:10.1007/s10637-020-01035-9 |
| [16] |
FU L L, CHEN S W, HE G, et al. Targeting extracellular signal-regulated protein kinase 1/2 (ERK1/2) in cancer: an update on pharmacological small-molecule inhibitors[J]. Journal of Medicinal Chemistry, 2022, 65(20): 13561-13573. DOI:10.1021/acs.jmedchem.2c01244 |
| [17] |
BHAGWAT S V, MCMILLEN W T, CAI S, et al. ERK inhibitor LY3214996 targets ERK pathway-driven cancers: a therapeutic approach toward precision medicine[J]. Molecular Cancer Therapeutics, 2020, 19(2): 325-336. DOI:10.1158/1535-7163.MCT-19-0183 |
| [18] |
LI A, JIAN S Z, YUAN X, et al. The ERK1/2 inhibitor, JSI-1187, demonstrates preclinical efficacy in tumor models with MAPK pathway mutations[J]. Cancer Research, 2020, 80(16): 4188-4189. |
| [19] |
PORTELINHA A, THOMPSON S, SMITH R A, et al. ASN007 is a selective ERK1/2 inhibitor with preferential activity against RAS- and RAF-mutant tumors[J]. Cell Reports Medicine, 2021, 2(7): 100350. DOI:10.1016/j.xcrm.2021.100350 |
| [20] |
MUNCK J M, BERDINI V, BEVAN L, et al. ASTX-029, a novel dual-mechanism ERK inhibitor, modulates both the phosphorylation and catalytic activity of ERK[J]. Molecular Cancer Therapeutics, 2021, 20(10): 1757-1768. DOI:10.1158/1535-7163.MCT-20-0909 |
| [21] |
WANG J M, QIN Y N, LIN M P, et al. Marine natural products from the Beibu Gulf: sources, chemistry, and bioactivities[J]. Marine Drugs, 2023, 21(2): 63. DOI:10.3390/md21020063 |
| [22] |
HEIGHTMAN T D, BERDINI V, BEVAN L, et al. Discovery of ASTX029, a clinical candidate which modulates the phosphorylation and catalytic activity of ERK1/2[J]. Journal of Medicinal Chemistry, 2021, 64(16): 12286-12303. DOI:10.1021/acs.jmedchem.1c00905 |
| [23] |
骆宜, 张乐, 王卫华, 等. 高效液相色谱-离子阱-飞行时间质谱鉴定决明子化学成分[J]. 药物分析杂志, 2015, 35(8): 1408-1416. |
| [24] |
张杰, 张振秋, 米宝丽, 等. 红镰霉素龙胆二糖苷和异红镰霉素龙胆二糖苷相互转化研究[J]. 中国药学杂志, 2014, 49(17): 1491-1495. |
| [25] |
NORMAN P. Evaluation of WO2012003387, Gilead's ASK1 inhibitors[J]. Expert Opinion on Therapeutic Patents, 2012, 22(4): 455-459. DOI:10.1517/13543776.2012.674515 |
| [26] |
LIU K, XIE Y, QIN Y, et al. The metal- and column-free synthesis of pyridin-2-yl ureas carrying cyclic secondary amine substituents and establishing the mechanism of the reactions through a computational study[J]. Organic Chemistry Frontiers, 2023, 10(13): 3182-3192. DOI:10.1039/D3QO00571B |
| [27] |
董玉洁, 蒋沅岐, 刘毅, 等. 决明子的化学成分、药理作用及质量标志物预测分析[J]. 中草药, 2021, 52(9): 2719-2732. |
| [28] |
ZHOU Y, XIANG S, YANG F, et al. Targeting gatekeeper mutations for kinase drug discovery[J]. Journal of Medicinal Chemistry, 2022, 65(23): 15540-15558. DOI:10.1021/acs.jmedchem.2c01361 |
| [29] |
ZHANG M, LIU Y, JANG H, et al. Strategy toward kinase-selective drug discovery[J]. Journal of Chemical Theory and Computation, 2023, 19(5): 1615-1628. DOI:10.1021/acs.jctc.2c01171 |
| [30] |
PAN X L, PEI J P, WANG A X, et al. Development of small molecule extracellular signal-regulated kinases (ERKs) inhibitors for cancer therapy[J]. Acta Pharmaceutica Sinica B, 2022, 12(5): 2171-2192. DOI:10.1016/j.apsb.2021.12.022 |
| [31] |
甘雨满, 刘永宏, 刘锴. 结合自由能微扰积分法及其在药物研发中的应用[J]. 广西科学, 2020, 27(5): 493-502. |